Human Protein Atlas in R
Laurent Gatto
de Duve Institute, UCLouvain, Belgiumhpar.RmdAbstract
The Human Protein Atlas (HPA) is a systematic study oh the human proteome using antibody-based proteomics. Multiple tissues and cell lines are systematically assayed affinity-purified antibodies and confocal microscopy. The hpar package is an R interface to the HPA project. It distributes three data sets, provides functionality to query these and to access detailed information pages, including confocal microscopy images available on the HPA web page.
Introduction
The HPA project
From the Human Protein Atlas (Uhlén et al. 2005; Uhlen et al. 2010) site:
The Swedish Human Protein Atlas project, funded by the Knut and Alice Wallenberg Foundation, has been set up to allow for a systematic exploration of the human proteome using Antibody-Based Proteomics. This is accomplished by combining high-throughput generation of affinity-purified antibodies with protein profiling in a multitude of tissues and cells assembled in tissue microarrays. Confocal microscopy analysis using human cell lines is performed for more detailed protein localisation. The program hosts the Human Protein Atlas portal with expression profiles of human proteins in tissues and cells.
The hpar package provides access to HPA data from the R interface. It also distributes the following data sets:
Several flat files are distributed by the HPA project and available within the package as data.frames, other datasets are available through a search query on the HPA website. The description below is taken from the HPA site:
hpaNormalTissue: Normal tissue data. Expression profiles for proteins in human tissues based on immunohistochemisty using tissue micro arrays. The tab-separated file includes Ensembl gene identifier (“Gene”), tissue name (“Tissue”), annotated cell type (“Cell type”), expression value (“Level”), and the gene reliability of the expression value (“Reliability”).
hpaNormalTissue16.1: Same as above, for version 16.1.
hpaCancer: Pathology data. Staining profiles for proteins in human tumor tissue based on immunohistochemisty using tissue micro arrays and log-rank P value for Kaplan-Meier analysis of correlation between mRNA expression level and patient survival. The tab-separated file includes Ensembl gene identifier (“Gene”), gene name (“Gene name”), tumor name (“Cancer”), the number of patients annotated for different staining levels (“High”, “Medium”, “Low” & “Not detected”) and log-rank p values for patient survival and mRNA correlation (“prognostic - favourable”, “unprognostic - favourable”, “prognostic - unfavourable”, “unprognostic - unfavourable”).
hpaCancer16.1: Same as above, for version 16.1
rnaConsensusTissue: RNA consensus tissue gene data. Consensus transcript expression levels summarized per gene in 54 tissues based on transcriptomics data from HPA and GTEx. The consensus normalized expression (“nTPM”) value is calculated as the maximum nTPM value for each gene in the two data sources. For tissues with multiple sub-tissues (brain regions, lymphoid tissues and intestine) the maximum of all sub-tissues is used for the tissue type. The tab-separated file includes Ensembl gene identifier (“Gene”), analysed sample (“Tissue”) and normalized expression (“nTPM”).
rnaHpaTissue: RNA HPA tissue gene data. Transcript expression levels summarized per gene in 256 tissues based on RNA-seq. The tab-separated file includes Ensembl gene identifier (“Gene”), analysed sample (“Tissue”), transcripts per million (“TPM”), protein-transcripts per million (“pTPM”) and normalized expression (“nTPM”).
rnaGtexTissue: RNA GTEx tissue gene data. Transcript expression levels summarized per gene in 37 tissues based on RNA-seq. The tab-separated file includes Ensembl gene identifier (“Gene”), analysed sample (“Tissue”), transcripts per million (“TPM”), protein-transcripts per million (“pTPM”) and normalized expression (“nTPM”). The data was obtained from GTEx.
rnaFantomTissue: RNA FANTOM tissue gene data. Transcript expression levels summarized per gene in 60 tissues based on CAGE data. The tab-separated file includes Ensembl gene identifier (“Gene”), analysed sample (“Tissue”), tags per million (“Tags per million”), scaled-tags per million (“Scaled tags per million”) and normalized expression (“nTPM”). The data was obtained from FANTOM5.
rnaGeneTissue21.0: RNA HPA tissue gene data. Transcript expression levels summarized per gene in 37 tissues based on RNA-seq, for hpa version 21.0. The tab-separated file includes Ensembl gene identifier (“Gene”), analysed sample (“Tissue”), transcripts per million (“TPM”), protein-transcripts per million (“pTPM”).
rnaGeneCellLine: RNA HPA cell line gene data. Transcript expression levels summarized per gene in 69 cell lines. The tab-separated file includes Ensembl gene identifier (“Gene”), analysed sample (“Cell line”), transcripts per million (“TPM”), protein-coding transcripts per million (“pTPM”) and normalized expression (“nTPM”).
rnaGeneCellLine16.1: Same as above, for version 16.1.
hpaSubcellularLoc: Subcellular location data. Subcellular location of proteins based on immunofluorescently stained cells. The tab-separated file includes the following columns: Ensembl gene identifier (“Gene”), name of gene (“Gene name”), gene reliability score (“Reliability”), enhanced locations (“Enhanced”), supported locations (“Supported”), Approved locations (“Approved”), uncertain locations (“Uncertain”), locations with single-cell variation in intensity (“Single-cell variation intensity”), locations with spatial single-cell variation (“Single-cell variation spatial”), locations with observed cell cycle dependency (type can be one or more of biological definition, custom data or correlation) (“Cell cycle dependency”), Gene Ontology Cellular Component term identifier (“GO id”).
hpaSubcellularLoc16.1: Same as above, for version 16.1.
hpaSubcellularLoc14: Same as above, for version 14.
hpaSecretome: Secretome data. The human secretome is here defined as all Ensembl genes with at least one predicted secreted transcript according to HPA predictions. The complete information about the HPA Secretome data is given on https://www.proteinatlas.org/humanproteome/blood/secretome. This dataset has 315 columns and includes the Ensembl gene identifier (“Gene”). Information about the additionnal variables can be found here by clicking on Show/hide columns.
The hpar::allHparData() returns a list of all datasets
(see below).
HPA data usage policy
The use of data and images from the HPA in publications and presentations is permitted provided that the following conditions are met:
- The publication and/or presentation are solely for informational and non-commercial purposes.
- The source of the data and/or image is referred to the HPA site1 and/or one or more of our publications are cited.
Installation
hpar is available through the Bioconductor project. Details about the package and the installation procedure can be found on its landing page. To install using the dedicated Bioconductor infrastructure, run :
## install BiocManager only one
install.packages("BiocManager")
## install hpar
BiocManager::install("hpar")After installation, hpar will have to be explicitly loaded with
library("hpar")## snapshotDate(): 2022-11-29## This is hpar version 1.41.1,
## based on the Human Protein Atlas
## Version: 21.1
## Release data: 2022.05.31
## Ensembl build: 103.38
## See '?hpar' or 'vignette('hpar')' for details.so that all the package’s functionality and data is available to the user.
The hpar package
Data sets
A table descibing all dataset available in the package can be
accessed with the allHparData() function.
hpa_data <- allHparData()The Title variable corresponds to names of the data that can be downloaded localled and cached as part of the ExperimentHub infrastructure.
head(normtissue <- hpaNormalTissue())## see ?hpar and browseVignettes('hpar') for documentation## loading from cache## Gene Gene.name Tissue Cell.type Level
## 1 ENSG00000000003 TSPAN6 adipose tissue adipocytes Not detected
## 2 ENSG00000000003 TSPAN6 adrenal gland glandular cells Not detected
## 3 ENSG00000000003 TSPAN6 appendix glandular cells Medium
## 4 ENSG00000000003 TSPAN6 appendix lymphoid tissue Not detected
## 5 ENSG00000000003 TSPAN6 bone marrow hematopoietic cells Not detected
## 6 ENSG00000000003 TSPAN6 breast adipocytes Not detected
## Reliability
## 1 Approved
## 2 Approved
## 3 Approved
## 4 Approved
## 5 Approved
## 6 ApprovedNote that given that the hpa data is distributed as par
the ExperimentHub infrastructure, it is also possible to query it
directly for relevant datasets.
library("ExperimentHub")
eh <- ExperimentHub()
query(eh, "hpar")## ExperimentHub with 15 records
## # snapshotDate(): 2022-11-29
## # $dataprovider: Human Protein Atlas
## # $species: Homo sapiens
## # $rdataclass: data.frame
## # additional mcols(): taxonomyid, genome, description,
## # coordinate_1_based, maintainer, rdatadateadded, preparerclass, tags,
## # rdatapath, sourceurl, sourcetype
## # retrieve records with, e.g., 'object[["EH7765"]]'
##
## title
## EH7765 | hpaCancer16.1
## EH7766 | hpaCancer
## EH7767 | hpaNormalTissue16.1
## EH7768 | hpaNormalTissue
## EH7769 | hpaSecretome
## ... ...
## EH7775 | rnaGeneCellLine16.1
## EH7776 | rnaGeneCellLine
## EH7777 | rnaGeneTissue21.0
## EH7778 | rnaGtexTissue
## EH7779 | rnaHpaTissueHPA interface
Each data described above is a data.frame and can be
easily manipulated using standard R or
BiocStyle::CRANpkg("tidyverse") tidyverse
functionality.
names(normtissue)## [1] "Gene" "Gene.name" "Tissue" "Cell.type" "Level"
## [6] "Reliability"## [1] 15323## [1] 141## [1] 63
## Number of genes highlighly and reliably expressed in each cell type
## in each tissue.
library("dplyr")
normtissue |>
filter(Reliability == "Approved",
Level == "High") |>
count(Cell.type, Tissue) |>
arrange(desc(n)) |>
head()## Cell.type Tissue n
## 1 glandular cells gallbladder 1578
## 2 glandular cells duodenum 1551
## 3 glandular cells small intestine 1532
## 4 glandular cells rectum 1490
## 5 glandular cells colon 1404
## 6 cells in tubules kidney 1357We will illustrate additional datasets using the TSPAN6 (tetraspanin 6) gene (ENSG00000000003) as example.
id <- "ENSG00000000003"
subcell <- hpaSubcellularLoc()## see ?hpar and browseVignettes('hpar') for documentation## loading from cache
rna <- rnaGeneCellLine()## see ?hpar and browseVignettes('hpar') for documentation
## loading from cache
## Compine protein immunohistochemisty data, with the subcellular
## location and RNA expression levels.
filter(normtissue, Gene == id) |>
full_join(filter(subcell, Gene == id)) |>
full_join(filter(rna, Gene == id)) |>
head()## Joining, by = c("Gene", "Gene.name", "Reliability")## Joining, by = c("Gene", "Gene.name")## Gene Gene.name Tissue Cell.type Level Reliability
## 1 ENSG00000000003 TSPAN6 adipose tissue adipocytes Not detected Approved
## 2 ENSG00000000003 TSPAN6 adipose tissue adipocytes Not detected Approved
## 3 ENSG00000000003 TSPAN6 adipose tissue adipocytes Not detected Approved
## 4 ENSG00000000003 TSPAN6 adipose tissue adipocytes Not detected Approved
## 5 ENSG00000000003 TSPAN6 adipose tissue adipocytes Not detected Approved
## 6 ENSG00000000003 TSPAN6 adipose tissue adipocytes Not detected Approved
## Main.location Additional.location Extracellular.location
## 1 Cell Junctions;Cytosol Nucleoli fibrillar center
## 2 Cell Junctions;Cytosol Nucleoli fibrillar center
## 3 Cell Junctions;Cytosol Nucleoli fibrillar center
## 4 Cell Junctions;Cytosol Nucleoli fibrillar center
## 5 Cell Junctions;Cytosol Nucleoli fibrillar center
## 6 Cell Junctions;Cytosol Nucleoli fibrillar center
## Enhanced Supported Approved Uncertain
## 1 Cell Junctions;Cytosol;Nucleoli fibrillar center
## 2 Cell Junctions;Cytosol;Nucleoli fibrillar center
## 3 Cell Junctions;Cytosol;Nucleoli fibrillar center
## 4 Cell Junctions;Cytosol;Nucleoli fibrillar center
## 5 Cell Junctions;Cytosol;Nucleoli fibrillar center
## 6 Cell Junctions;Cytosol;Nucleoli fibrillar center
## Single.cell.variation.intensity Single.cell.variation.spatial
## 1 Cytosol
## 2 Cytosol
## 3 Cytosol
## 4 Cytosol
## 5 Cytosol
## 6 Cytosol
## Cell.cycle.dependency
## 1
## 2
## 3
## 4
## 5
## 6
## GO.id
## 1 Cell Junctions (GO:0030054);Cytosol (GO:0005829);Nucleoli fibrillar center (GO:0001650)
## 2 Cell Junctions (GO:0030054);Cytosol (GO:0005829);Nucleoli fibrillar center (GO:0001650)
## 3 Cell Junctions (GO:0030054);Cytosol (GO:0005829);Nucleoli fibrillar center (GO:0001650)
## 4 Cell Junctions (GO:0030054);Cytosol (GO:0005829);Nucleoli fibrillar center (GO:0001650)
## 5 Cell Junctions (GO:0030054);Cytosol (GO:0005829);Nucleoli fibrillar center (GO:0001650)
## 6 Cell Junctions (GO:0030054);Cytosol (GO:0005829);Nucleoli fibrillar center (GO:0001650)
## Cell.line TPM pTPM nTPM
## 1 A-431 21.3 25.6 24.8
## 2 A549 20.5 24.4 23.0
## 3 AF22 78.8 96.9 80.6
## 4 AN3-CA 38.7 47.1 42.2
## 5 ASC diff 19.2 22.1 28.7
## 6 ASC TERT1 11.3 13.1 16.6It is also possible to directly open the HPA page for a specific gene (see figure below).
browseHPA(id)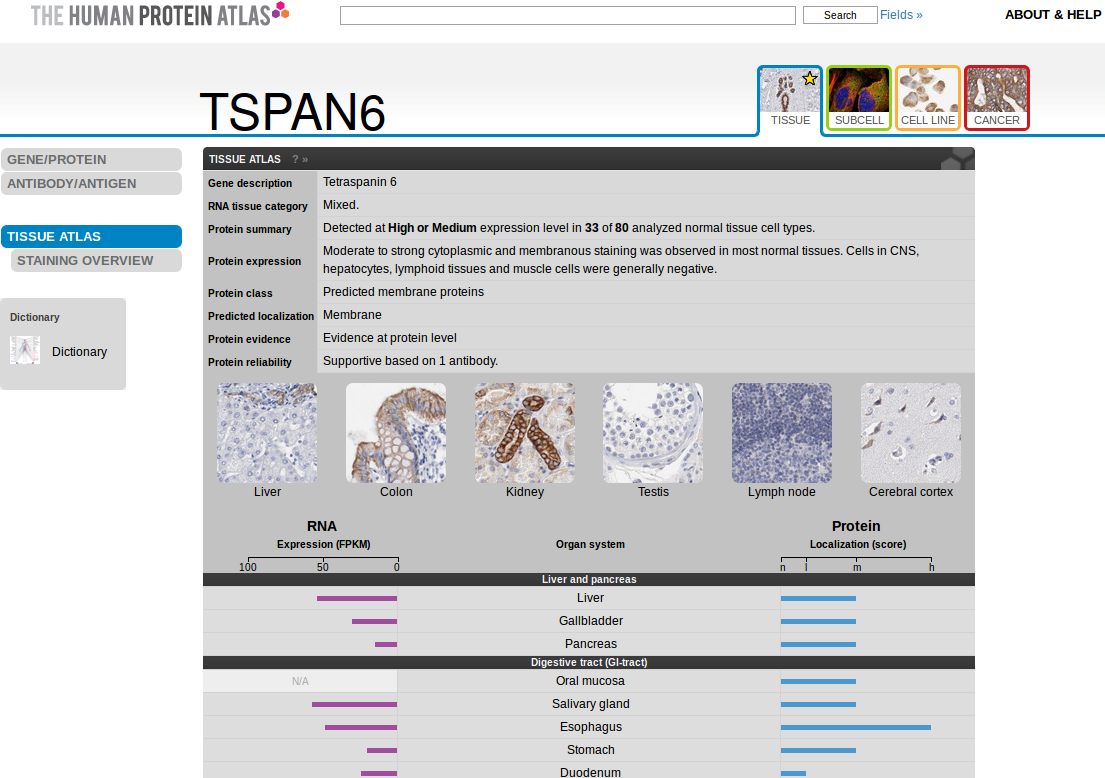
The HPA web page for the tetraspanin 6 gene (ENSG00000000003).
HPA release information
Information about the HPA release used to build the installed hpar package
can be accessed with getHpaVersion, getHpaDate
and getHpaEnsembl. Full release details can be found on the
HPA release
history page.
## version
## "21.1"## date
## "2022.05.31"## ensembl
## "103.38"A small use case
Let’s compare the subcellular localisation annotation obtained from the HPA subcellular location data set and the information available in the Bioconductor annotation packages.
id <- "ENSG00000001460"
filter(subcell, Gene == id)## Gene Gene.name Reliability Main.location Additional.location
## 1 ENSG00000001460 STPG1 Approved Nucleoplasm
## Extracellular.location Enhanced Supported Approved Uncertain
## 1 Nucleoplasm
## Single.cell.variation.intensity Single.cell.variation.spatial
## 1
## Cell.cycle.dependency GO.id
## 1 Nucleoplasm (GO:0005654)Below, we first extract all cellular component GO terms available for
id from the org.Hs.eg.db
human annotation and then retrieve their term definitions using the
GO.db
database.
library("org.Hs.eg.db")
library("GO.db")
ans <- AnnotationDbi::select(org.Hs.eg.db, keys = id,
columns = c("ENSEMBL", "GO", "ONTOLOGY"),
keytype = "ENSEMBL")## 'select()' returned 1:many mapping between keys and columns
ans <- ans[ans$ONTOLOGY == "CC", ]
ans## ENSEMBL GO EVIDENCE ONTOLOGY
## 2 ENSG00000001460 GO:0005634 IEA CC
## 3 ENSG00000001460 GO:0005739 IEA CC## GO:0005634 GO:0005739
## "nucleus" "mitochondrion"Session information
## R Under development (unstable) (2022-11-23 r83380)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 22.04.1 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] ExperimentHub_2.7.0 AnnotationHub_3.7.0 BiocFileCache_2.7.0
## [4] dbplyr_2.2.1 hpar_1.41.1 GO.db_3.16.0
## [7] org.Hs.eg.db_3.16.0 AnnotationDbi_1.61.0 IRanges_2.33.0
## [10] S4Vectors_0.37.0 Biobase_2.59.0 BiocGenerics_0.45.0
## [13] dplyr_1.0.10 BiocStyle_2.27.0
##
## loaded via a namespace (and not attached):
## [1] tidyselect_1.2.0 blob_1.2.3
## [3] filelock_1.0.2 Biostrings_2.67.0
## [5] bitops_1.0-7 fastmap_1.1.0
## [7] RCurl_1.98-1.9 promises_1.2.0.1
## [9] digest_0.6.30 mime_0.12
## [11] lifecycle_1.0.3 ellipsis_0.3.2
## [13] KEGGREST_1.39.0 interactiveDisplayBase_1.37.0
## [15] RSQLite_2.2.19 magrittr_2.0.3
## [17] compiler_4.3.0 rlang_1.0.6
## [19] sass_0.4.4 tools_4.3.0
## [21] utf8_1.2.2 yaml_2.3.6
## [23] knitr_1.41 htmlwidgets_1.5.4
## [25] bit_4.0.5 curl_4.3.3
## [27] withr_2.5.0 purrr_0.3.5
## [29] desc_1.4.2 fansi_1.0.3
## [31] xtable_1.8-4 cli_3.4.1
## [33] rmarkdown_2.18 crayon_1.5.2
## [35] ragg_1.2.4 generics_0.1.3
## [37] httr_1.4.4 DBI_1.1.3
## [39] cachem_1.0.6 stringr_1.4.1
## [41] zlibbioc_1.45.0 assertthat_0.2.1
## [43] BiocManager_1.30.19 XVector_0.39.0
## [45] vctrs_0.5.1 jsonlite_1.8.3
## [47] bookdown_0.30 bit64_4.0.5
## [49] systemfonts_1.0.4 crosstalk_1.2.0
## [51] jquerylib_0.1.4 glue_1.6.2
## [53] pkgdown_2.0.6.9000 DT_0.26
## [55] stringi_1.7.8 BiocVersion_3.17.1
## [57] later_1.3.0 GenomeInfoDb_1.35.5
## [59] tibble_3.1.8 pillar_1.8.1
## [61] rappdirs_0.3.3 htmltools_0.5.3
## [63] GenomeInfoDbData_1.2.9 R6_2.5.1
## [65] textshaping_0.3.6 rprojroot_2.0.3
## [67] evaluate_0.18 shiny_1.7.3
## [69] png_0.1-8 memoise_2.0.1
## [71] httpuv_1.6.6 bslib_0.4.1
## [73] Rcpp_1.0.9 xfun_0.35
## [75] fs_1.5.2 pkgconfig_2.0.3www.proteinatlas.org↩︎