
MSnbase contributors 2010 - 2016 2017
MSnbase (or here on GitHub) (Gatto and Lilley, 2010) is one of my main software projects. I started working on the package when I moved to Cambridge in 2010. It offers a way to import, manipulate and process raw mass spectrometry and quantitative proteomics data in R.
Since then, I benefited from quite a few contributions (which I already briefly highlighted here). In this post, I want to give a few more details about and credit to the contributors.
The figure below summarises the contributors over time.
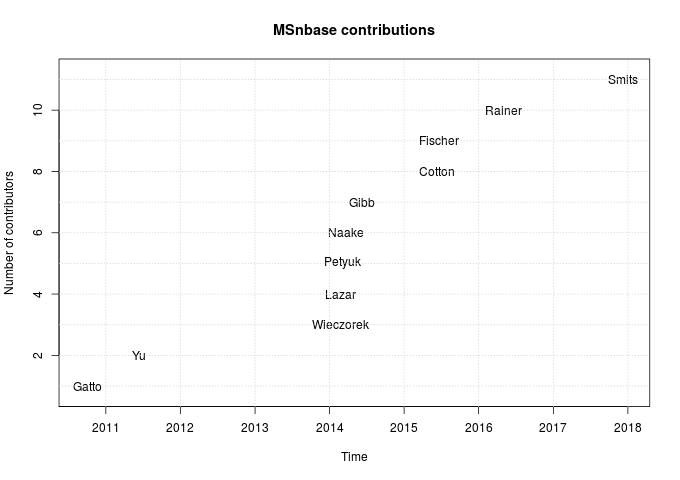
The first data point on the plot is Mon Oct 4 23:35:23 2010, and corresponds to the very first git commit (with typo) in the GitHub repository:
commit 7cb6b1a598d0b2ed55234f75229b925ceb26afaa
Author: Laurent Gatto <laurent.gatto@gmail.com>
Date: Mon Oct 4 23:35:23 2010 +0100
Inital commit - package dir structure and R code
All the R code files, some with roxygen incode documentation
as well as the R package structure are committed. Included
are also DESCRIPTION, NAMESPACE, NEWS (empty) and README.org.
A dataset in mzXML format, dummyiTRAQ.mzXML in inst/extdata
will serve as testing file.Since then, the commits have been quite regular, except for 2012.
Guangchuang Yu
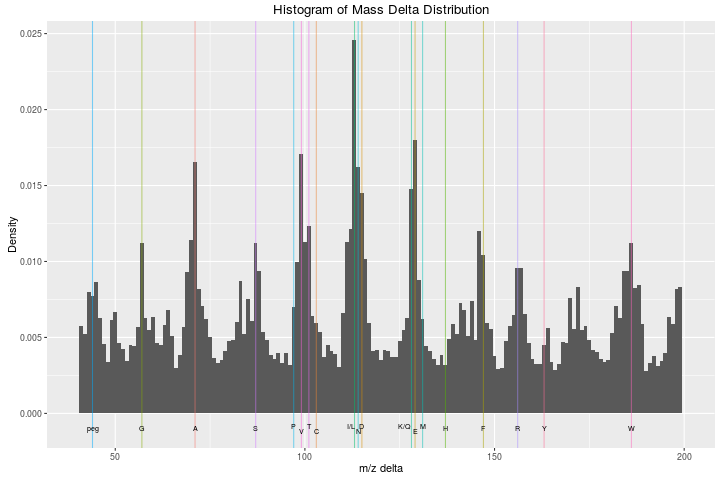
Guangchuang contributed the plotMzDelta function in June 2011. The
function produces a figure used as a quality for MS2 spectra, as
detailed in
Foster et al. 2011. All
differences between neighbouring peaks in MS2 spectra are calculated
and plotted as a histogram. Assuming good peptide fragmentation and
absence of contamination, the histogram should feature peaks
corresponding to amino acids.
He is also the author of the first implementation of the readMgfData
that, as the name implies, reads mgf data (thanks for the
reminder).
Samuel Wieczorek and Cosmin Lazar
The contributions of Sam and Cosmin in February 2014 stem from our
work on missing value imputation in quantitative proteomics
(Lazar et al. 2016)
and have materialised in improvements in the impute function.
Vlad Petyuk
Vlad’s main contrition was in the combineFeatures function that
aggregates low level features, in March 2014. He contributed the
redundancy handler, that defines how to handle peptides that can be
associated to multiple higher-level features (proteins).
The ggplot2-based implementation of image, that produces a simple
heatmap of the quantitative data also come from him, and is based on
his own vp.misc::image_msnset implementation.
Thomas Naake
Thomas visited the group as an Erasmus student from April to June 2014
and implemented the first version of the
pRolocGUI
package (and
here on
GitHub). During this work, we discussed about features that would be
needed for the interactive visualisation that ended up being
implemented/added to MSnbase and then used in the GUIs. The main one
I can remember is the FreaturesOfInterest class, that stores an
arbitrary set of features (proteins) that can then conveniently
highlighted on a PCA plot using the highlightOnPlot function from
the pRoloc package.
library("pRolocdata")
data("tan2009r1")
x <- FeaturesOfInterest(description = "A test set of features of interest",
fnames = featureNames(tan2009r1)[1:10],
object = tan2009r1)
plot2D(tan2009r1)
highlightOnPlot(tan2009r1, x)
highlightOnPlot(tan2009r1, x, labels = TRUE, pos = 3)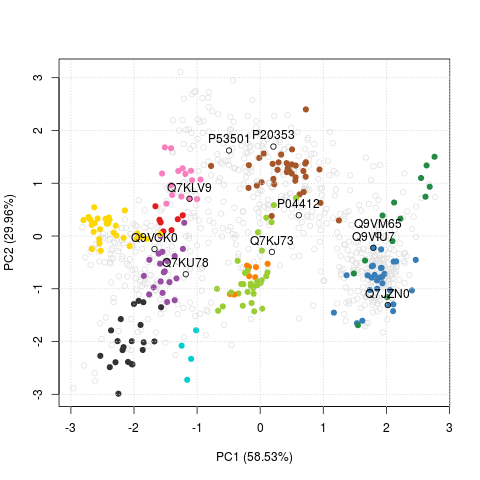
Sebastian Gibb
Sebastian visited the group for 3 months in 2014. He did a lot of work
on MSnbase and
synapter
(here) and is still
active. Among his many contributions are the addIdentificationData,
that adds identification data from mzid files to raw (MSnExp
objects) and quantitative (MSnSet objects) data. He also added
various raw data processing functions (such as smoothing, peak
picking) by leveraging existing code in his
MALDIQuant
package and support for label-free MS2 quantitation. He also worked on
spectra comparison, annotation and visualisation, as illustrated
below.
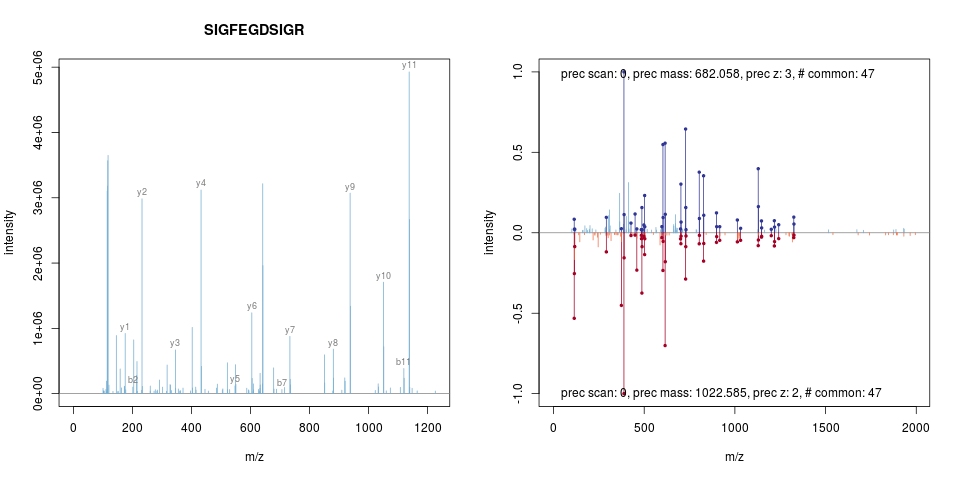
Richie Cotton
Richie contributed in supporting mzTab version 1.0, as described in
issue #41 from
June 2015. I updated his code to fit into the MSnbase infrastructure
and annotate some the ontology controlled parameters (using the
rols
package).
Martina Fischer
In June 2015, Martina contributed a whole new method for feature
aggregation, termed iPQF (for Isobaric Protein Quantification based
on Features)). iPQF is a new peptide-to-protein summarisation method
using peptide spectra characteristics to improve protein
quantification. All details in
Fischer and Renard, 2016,
readily available using combineFeatures(..., method = "iPQF").
Johannes Rainer
Johannes has been instrumental in the recent (October 2016) release of
MSnbase version 2.0. During summer 2016, we worked on a new backend
for raw data. Instead of loading spectra into memory, as in the
original MSnExp implementation, the alternative implementation
accesses the raw data from the hard drive on-the-fly only when it is
needed. This is made possible by the fast on-disk access provided by
the
mzR
package (here on GitHub) that uses
the proteowizard C/C++ code
base under the hood. For more details and a direct comparison, see the
benchmarking
vignette.
This joint work with Johannes aims at providing a common and efficient infrastructure for mass spectrometry data that can be used by the proteomics and metabolomics developers.
Arne Smits
Arne developes the
DEP
package for differential enrichment analysis of proteomics data and
has contributed MSnSet
to/from SummarizedExperiment methods, to facilitate the
inter-operability between his package and MSnbase.
Seeing that MSnbase is used and attracts attention from other
developers is a great reward for me. Thank you all for your valuable
contributions!
Updates Since its first publication (2016-11-27), this post has been ammended to add Arne Smits’ contribution (2018-01-04).
