
Notes on the CZI workshop on single cell proteomics and metabolomics
Here’s a summary of my recent short intro talk for the recent virtual CZI workshop on single cell proteomics and metabolomics, 8 April 2022.

Overview
As an example of state-of-the-art infrastructure, I would like to present the R for Mass Spectrometry initiative and illustrate some of our approaches to tackle software and analysis related bottlenecks in mass spectrometry-based proteomics and metabolomics.
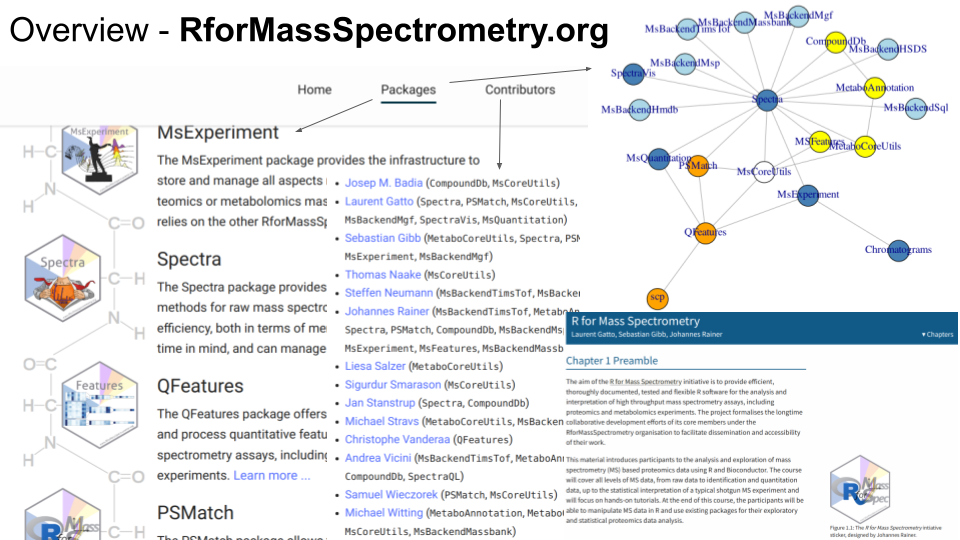
R for Mass Spectrometry is formed by over 20 tightly integrated and interoperable software packages, tackling the whole spectrum of data and their analyses, from raw mass spectrometry data, peptide and metabolite identification, quantitative data analysis, up to single-cell proteomics data processing. Each package is thoroughly tested, documented and peer-reviewed. Once a package reaches maturity, it get submitted to and reviewed by the Bioconductor project. There’s a project-level web page in addition to each package’s specific page and a book/tutorial.
There are currently 14 contributors. The 3 main developers and initiators of the project have over 30 years of cumulative experience in software development and data analysis in mass spectrometry-based proteomics and metabolomics.
A trend worth highlighting is the move away from large, monolithic, one-size-fits-all software (especially important for fast-evolving technologies that require flexibility) and the integration/re-unification of these individual components/tools at the workflow level.
Bottlenecks
I would like to mention a couple of high-level challenges, beyond those that are at our reach, such as the handling of missing data (be it through technological means such as DIA, software solution, and/or both), or batch effect (through better experimental designs and/or software means) and the interaction between these two (see here).
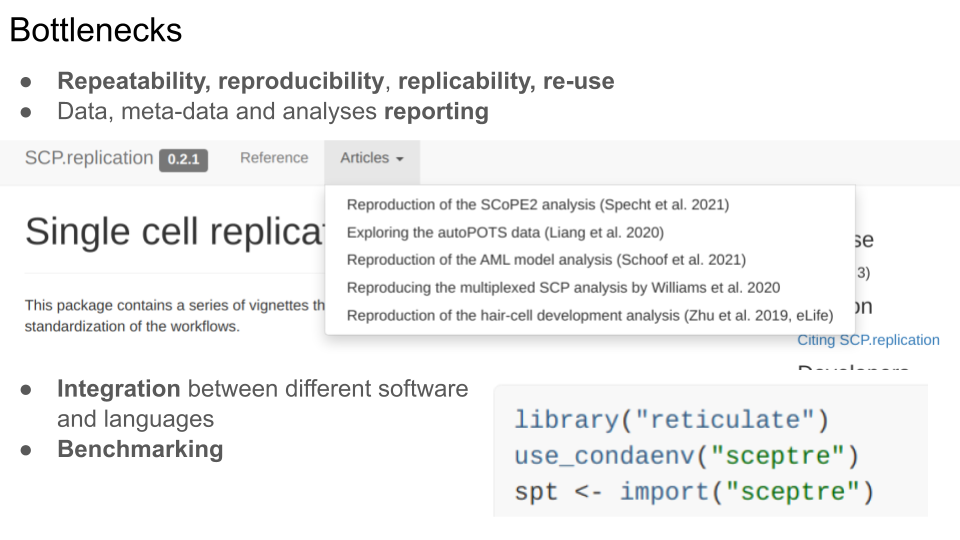
- Repeatability, reproducibility, replicability and re-use: we are still struggling at the whole range of “doing things again”, be it with one’s own data some time later, re-analysing someone’s else data, using either the exact software or different ones.
- This struggle is in great part related to poor data, meta-data and analysis reporting.
These are particularly problematic at an early stage, when a technology and its applications are developed and mature, such as single-cell proteomics/metabolomics. The example of the SCP.replication package illustrates the reproduction and replication of some published studies, some of which we were able to reproduce and replicate, others not at all (due for example to wrong supplementary files, missing information about how the data were analysed, or completely missing meta-data).
-
The need for integration between software and languages is inevitable - it provides access to more tools, even if at the cost of increased complexity in code. Here’s short example from one of the replications mentioned above, where we use reticulate to call the SCeptre Python package. Julia is another language that provides unique libraries and features, that can be extremely useful for the analysis of multi-condition single cell data.
-
Some of these challenges and bottlenecks could be addressed, at least to some extend, through better benchmarking. Benchmarking is however a very difficult exercise that also required better reporting of data, meta-data and analyses details.
Opportunities
In terms of community, I think that the CZI Single Cell Biology Program has demonstrated some notable successes. Related to some of the more technical points I have mentioned above, interoperability between the R and Python ecosystems of single-cell RNA sequencing and common data structures have emerged.

From a software point of view, the Essential Open Source Software for Science has gotten a lot of visibility in genomics, less so in proteomics and metabolomics. I see an opportunity to aim for a common corpus of re-usable and interoperable software and methods as well as better integration between software across platforms and operating systems. The SpectriPy/ is one effort in the R for Mass Spectrometry initiative.
I mentioned repeatability, reproducibility, replicability and better reporting. We need better benchmarking, not necessarily as a way to identify “the best software” (which is hardly the best, by merely the one that performed better in a given data/user setting), but more as a means to integrate and unify common experiences and expertise into a workflow to address more difficult tasks.
In terms of single cell biology, we have a lot to learn and be inspired by what in done in genomics, including their experience in data integration. But in terms of applications, it’s important to differentiate from genomics. I don’t think that aiming for the same application, or at least no only or mainly the definition of cell types and states (including dimensionality reduction, clustering, …) is the best approach. Indeed, we currently lack scalability (i.e. tens, hundreds of thousands or even millions of cells) and it will be difficult to reach the level of genomics at a sensible cost. We should focus for example on the inference of regulatory interactions with minimal assumptions, notable in the light of post translational modifications. And to do this, we need open and well-annotated data and software.
