A transfer learning algorithm for spatial proteomics
Lisa M. Breckels
Computational Proteomics Unit, Cambridge, UKLaurent Gatto
de Duve Institute, UCLouvain, BelgiumSource:
vignettes/v05-pRoloc-transfer-learning.Rmd
v05-pRoloc-transfer-learning.RmdAbstract
This vignette illustrates the application of a transfer learning algorithm to assign proteins to sub-cellular localisations. The knntlClassification algorithm combines primary experimental spatial proteomics data (LOPIT, PCP, etc.) and an auxiliary data set (for example binary data based on Gene Ontology terms) to improve the sub-cellular assignment given an optimal combination of these data sources.
Introduction
Our main data source to study protein sub-cellular localisation are high-throughput mass spectrometry-based experiments such as LOPIT, PCP and similar designs (see (Gatto et al. 2010) for an general introduction). Recent optimised experiments result in high quality data enabling the identification of over 6000 proteins and discriminate numerous sub-cellular and sub-organellar niches (Christoforou et al. 2016). Supervised and semi-supervised machine learning algorithms can be applied to assign thousands of proteins to annotated sub-cellular niches (Lisa M. Breckels et al. 2013, Gatto:2014) (see also the pRoloc-tutorial vignette). These data constitute our main source for protein localisation and are termed thereafter primary data.
There are other sources of data about sub-cellular localisation of proteins, such as the Gene Ontology (Ashburner et al. 2000) (in particular the cellular compartment name space), quantitative features derived from protein sequences (such as pseudo amino acid composition) or the Human Protein Atlas (Uhlen et al. 2010) to cite a few. These data, while not optimised to a specific system at hand and, in the case of annotation feature, not as reliable as our experimental data, constitute an invaluable, often plentiful source of auxiliary information.
The aim of a transfer learning algorithm is to combine different sources of data to improve overall classification. In particular, the goal is to support/complement the primary target domain (experimental data) with auxiliary data (annotation) features without compromising the integrity of our primary data. In this vignette, we describe the application of transfer learning algorithms for the localisation of proteins from the pRoloc package, as described in
Breckels LM, Holden S, Wonjar D, Mulvey CM, Christoforou A, Groen A, Trotter MW, Kohlbacker O, Lilley KS and Gatto L (2016). Learning from heterogeneous data sources: an application in spatial proteomics. PLoS Comput Biol 13;12(5):e1004920. doi: 10.1371/journal.pcbi.1004920.
Two algorithms were developed: a transfer learning algorithm based on the -nearest neighbour classifier, coined kNN-TL hereafter, described in section @ref(sec:knntl), and one based on the support vector machine algorithm, termed SVM-TL, described in section @ref(sec:svmtl).
Preparing the auxiliary data
The Gene Ontology
The auxiliary data is prepared from the primary data’s features. All the GO terms associated to these features are retrieved and used to create a binary matrix where a one (zero) at position indicates that term has (not) been used to annotate feature .
The GO terms are retrieved from an appropriate repository using the biomaRt package. The specific Biomart repository and query will depend on the species under study and the type of features. The first step is to prepare annotation parameters that will enable to perform the query. The pRoloc package provides a dedicated infrastructure to set up the query to the annotation resource and prepare the GO data for subsequent analyses. This infrastructure is composed of:
- define the annotation parameters based on the species and feature types;
- query the resource defined in (1) to retrieve relevant terms and use the terms to prepare the auxiliary data.
We will demonstrate these steps using a LOPIT experiment on Human
Embryonic Kidney (HEK293T) fibroblast cells (Lisa
M. Breckels et al. 2013), available and documented in the pRolocdata
experiment package as andy2011.
library("pRolocdata")
data(andy2011)Preparing the query parameters
The query parameters are stored as AnnotationParams objects that are created with the setAnnotationParams function. The function will present a first menu with 486. Once the species has been selected, a set of possible identifier types is displayed.
AnnotationParams instance for the
human andy2011 data.It is also possible to pass patterns to match against the species
("Human genes") and feature type
("UniProtKB/Swiss-prot ID").
ap <- setAnnotationParams(inputs =
c("Human genes",
"UniProtKB/Swiss-Prot ID"))## Using species Human genes (GRCh38.p13)## Warning: Ensembl will soon enforce the use of https.
## Ensure the 'host' argument includes "https://"## Using feature type UniProtKB/Swiss-Prot ID(s) [e.g. A0A024R1R8]## Connecting to Biomart...## Warning: Ensembl will soon enforce the use of https.
## Ensure the 'host' argument includes "https://"
ap## Object of class "AnnotationParams"
## Using the 'ENSEMBL_MART_ENSEMBL' BioMart database
## Using the 'hsapiens_gene_ensembl' dataset
## Using 'uniprotswissprot' as filter
## Created on Sat Nov 23 16:07:23 2024The setAnnotationParams function automatically sets the
annotation parameters globally so that the ap object does
not need to be explicitly set later on. The default parameters can be
retrieved with getAnnotationParams.
Preparing the auxiliary data from the GO ontology
The feature names of the andy2011 data are UniProt
identifiers, as defined in the ap accession parameters.
data(andy2011)
head(featureNames(andy2011))## [1] "O00767" "P51648" "Q2TAA5" "Q9UKV5" "Q12797" "P16615"The makeGoSet function takes an MSnSet class (from
which the feature names will be extracted) or, directly a vector of
characters containing the feature names of interest to retrieve the
associated GO terms and construct an auxiliary MSnSet. By
default, it downloads cellular component terms and does not do
any filtering on the terms evidence codes (see the makeGoSet
manual for details). Unless passed as argument, the default, globally
set AnnotationParams are used to define the Biomart server and
the query.
andygoset <- makeGoSet(andy2011)
andygoset## MSnSet (storageMode: lockedEnvironment)
## assayData: 1371 features, 918 samples
## element names: exprs
## protocolData: none
## phenoData: none
## featureData
## featureNames: O00767 P51648 ... O75312 (1371 total)
## fvarLabels: Accession.No. Protein.Description ...
## UniProtKB.entry.name (10 total)
## fvarMetadata: labelDescription
## experimentData: use 'experimentData(object)'
## Annotation:
## - - - Processing information - - -
## Constructed GO set using cellular_component namespace [Sat Nov 23 16:07:25 2024]
## MSnbase version: 2.33.2
exprs(andygoset)[1:7, 1:4]## GO:0016020 GO:0005783 GO:0005789 GO:0005730
## O00767 1 1 1 1
## P51648 1 1 1 0
## Q2TAA5 1 1 1 0
## Q9UKV5 1 1 1 0
## Q12797 1 1 1 0
## P16615 1 1 1 0
## Q96SQ9 1 1 1 0We now have a primary data set, composed of 1371 protein quantitative profiles for 8 fractions along the density gradient and an auxiliary data set for 918 cellular compartment GO terms for the same 1371 features.
A note on reproducibility
The generation of the auxiliary data relies on specific Biomart server Mart instances in the AnnotationParams class and the actual query to the server to obtain the GO terms associated with the features. The utilisation of online servers, which undergo regular updates, does not guarantee reproducibility of feature/term association over time. It is recommended to save and store the AnnotationParams and auxiliary MSnSet instances. Alternatively, it is possible to use other Bioconductor infrastructure, such as specific organism annotations and the GO.db package to use specific versioned (and thus traceable) annotations.
The Human Protein Atlas
The feature names of our LOPIT experiment are UniProt identifiers,
while the Human Protein Atlas uses Ensembl gene identifiers. This first
code chunk matches both identifier types using the Ensembl Biomart
server and left_join from the dplyr package.
In this section, we copy the experimental data to
andyhpa.
andyhpa <- andy2011
fvarLabels(andyhpa)[1] <- "accession" ## for left_join matching
## convert protein accession numbers to ensembl gene identifiers
library("biomaRt")
mart <- useMart("ensembl", dataset = "hsapiens_gene_ensembl")
filter <- "uniprotswissprot"
attrib <- c("uniprot_gn_symbol", "uniprotswissprot", "ensembl_gene_id")
bm <- getBM(attributes = attrib,
filters = filter,
values = fData(andyhpa)[, "accession"],
mart = mart)
head(bm)## uniprot_gn_symbol uniprotswissprot ensembl_gene_id
## 1 ESYT2 A0FGR8 ENSG00000117868
## 2 ILVBL A1L0T0 ENSG00000105135
## 3 NBAS A2RRP1 ENSG00000151779
## 4 MTMR11 A4FU01 ENSG00000014914
## 5 C2orf72 A6NCS6 ENSG00000204128
## 6 RTL1 A6NKG5 ENSG00000254656
## HPA data
library("hpar")## This is hpar version 1.49.0,
## based on the Human Protein Atlas
## Version: 21.1
## Release data: 2022.05.31
## Ensembl build: 103.38
## See '?hpar' or 'vignette('hpar')' for details.
## using old version for traceability
hpa <- hpar::hpaSubcellularLoc14()## see ?hpar and browseVignettes('hpar') for documentation## loading from cache
hpa$Reliability <- droplevels(hpa$Reliability)
colnames(hpa)[1] <- "ensembl_gene_id"
hpa <- dplyr::left_join(hpa, bm)## Joining with `by = join_by(ensembl_gene_id)`
hpa <- hpa[!duplicated(hpa$uniprotswissprot), ]
## match HPA/LOPIT
colnames(hpa)[7] <- "accession"
fd <- dplyr::left_join(fData(andyhpa), hpa)## Joining with `by = join_by(accession)`
rownames(fd) <- featureNames(andyhpa)
fData(andyhpa) <- fd
stopifnot(validObject(andyhpa))
## Let's get rid of features without any hpa data
lopit <- andyhpa[!is.na(fData(andyhpa)$Main.location), ]Below, we deparse the multiple ‘;’-delimited locations contained in the Human Protein sub-cellular Atlas, create the auxiliary binary data matrix (only localisations with reliability equal to Supportive are considered; Uncertain assignments are ignored - see http://www.proteinatlas.org/about/antibody+validation for details) and filter proteins without any localisation data.
## HPA localisation
hpalocs <- c(as.character(fData(lopit)$Main.location),
as.character(fData(lopit)$Other.location))
hpalocs <- hpalocs[!is.na(hpalocs)]
hpalocs <- unique(unlist(strsplit(hpalocs, ";")))
makeHpaSet <- function(x, score2, locs = hpalocs) {
hpamat <- matrix(0, ncol = length(locs), nrow = nrow(x))
colnames(hpamat) <- locs
rownames(hpamat) <- featureNames(x)
for (i in 1:nrow(hpamat)) {
loc <- unlist(strsplit(as.character(fData(x)[i, "Main.location"]), ";"))
loc2 <- unlist(strsplit(as.character(fData(x)[i, "Other.location"]), ";"))
score <- score2[as.character(fData(x)[i, "Reliability"])]
hpamat[i, loc] <- score
hpamat[i, loc2] <- score
}
new("MSnSet", exprs = hpamat,
featureData = featureData(x))
}
hpaset <- makeHpaSet(lopit,
score2 = c(Supportive = 1, Uncertain = 0))
hpaset <- filterZeroRows(hpaset)## Removing 318 columns with only 0s.
dim(hpaset)## [1] 669 18## Endoplasmic reticulum Cytoplasm Vesicles
## O00767 1 0 0
## O95302 0 0 1
## P10809 0 0 0Protein-protein interactions
Protein-protein interaction data can also be used as auxiliary data input to the transfer learning algorithm. Several sources can be used to do so directly from R:
The PSICQUIC package provides an R interfaces to the HUPO Proteomics Standard Initiative (HUPO-PSI) project, which standardises programmatic access to molecular interaction databases. This approach enables to query great many resources in one go but, as noted in the vignettes, for bulk interactions, it is recommended to directly download databases from individual PSICQUIC providers.
The STRINGdb package provides a direct interface to the STRING protein-protein interactions database. This package can be used to generate a table as the one used below. The exact procedure is described in the
STRINGdbvignette and involves mapping the protein identifiers with the map and retrieve the interaction partners with the get_neighbors method.Finally, it is possible to use any third-party PPI inference results and adequately prepare these results for transfer learning. Below, we will described this case with PPI data in a tab-delimited format, as retrieved directly from the STRING database.
Below, we access the PPI spreadsheet file for our test data, that is distributed with the pRolocdata package.
ppif <- system.file("extdata/tabdelimited._gHentss2F9k.txt.gz", package = "pRolocdata")
ppidf <- read.delim(ppif, header = TRUE, stringsAsFactors = FALSE)
head(ppidf)## X.node1 node2 node1_string_id node2_string_id node1_external_id
## 1 NUDT5 IMPDH2 1861432 1850365 ENSP00000419628
## 2 NOP2 RPL23 1858730 1858184 ENSP00000382392
## 3 HSPA4 ENO1 1848476 1843405 ENSP00000302961
## 4 RPS13 DKC1 1862013 1855055 ENSP00000435777
## 5 RPL35A DDOST 1859718 1856225 ENSP00000393393
## 6 RPL13A RPS6 1857955 1857216 ENSP00000375730
## node2_external_id neighborhood fusion cooccurence homology coexpression
## 1 ENSP00000321584 0.000 0 0 0 0.112
## 2 ENSP00000377865 0.000 0 0 0 0.064
## 3 ENSP00000234590 0.000 0 0 0 0.109
## 4 ENSP00000358563 0.462 0 0 0 0.202
## 5 ENSP00000364188 0.000 0 0 0 0.000
## 6 ENSP00000369757 0.000 0 0 0 0.931
## experimental knowledge textmining combined_score
## 1 0.000 0.0 0.370 0.416
## 2 0.868 0.0 0.000 0.871
## 3 0.222 0.0 0.436 0.575
## 4 0.000 0.0 0.354 0.698
## 5 0.000 0.9 0.265 0.923
## 6 0.419 0.9 0.240 0.996The file contains 18623 pairwise interactions and the STRING combined interaction score. Below, we create a contingency matrix that uses these scores to encode and weight interactions.
uid <- unique(c(ppidf$X.node1, ppidf$node2))
ppim <- diag(length(uid))
colnames(ppim) <- rownames(ppim) <- uid
for (k in 1:nrow(ppidf)) {
i <- ppidf[[k, "X.node1"]]
j <- ppidf[[k, "node2"]]
ppim[i, j] <- ppidf[[k, "combined_score"]]
}
ppim[1:5, 1:8]## NUDT5 NOP2 HSPA4 RPS13 RPL35A RPL13A CPS1 CTNNB1
## NUDT5 1 0 0 0 0.000 0.000 0 0
## NOP2 0 1 0 0 0.000 0.000 0 0
## HSPA4 0 0 1 0 0.000 0.000 0 0
## RPS13 0 0 0 1 0.997 0.998 0 0
## RPL35A 0 0 0 0 1.000 0.999 0 0We now have a contingency matrix reflecting a total of 19910
interactions between 1287 proteins. Below, we only keep proteins that
are also available in our spatial proteomics data (renamed to
andyppi), subset the PPI and LOPIT data, create the
appropriate MSnSet object, and filter out proteins without
any interaction scores.
andyppi <- andy2011
featureNames(andyppi) <- sub("_HUMAN", "", fData(andyppi)$UniProtKB.entry.name)
cmn <- intersect(featureNames(andyppi), rownames(ppim))
ppim <- ppim[cmn, ]
andyppi <- andyppi[cmn, ]
ppi <- MSnSet(ppim, fData = fData(andyppi),
pData = data.frame(row.names = colnames(ppim)))
ppi <- filterZeroCols(ppi)## Removing 178 columns with only 0s.We now have two MSnSet objects containing respectively
520 primary experimental protein profiles along a sub-cellular density
gradient (andyppi) and 520 auxiliary interaction profiles
(ppi).
Support vector machine transfer learning
The SVM-TL method descibed in (L. M. Breckels et al. 2016) has not yet been incorporated in the pRoloc package. The code implementing the method is currently available in its own repository:
https://github.com/ComputationalProteomicsUnit/lpsvm-tl-code
Nearest neighbour transfer learning
Optimal weights
The weighted nearest neighbours transfer learning algorithm estimates
optimal weights for the different data sources and the spatial niches
described for the data at hand with the knntlOptimisation
function. For instance, for the human data modelled by the
andy2011 and andygoset objects1 and the 10 annotated
sub-cellular localisations (Golgi, Mitochondrion, PM, Lysosome, Cytosol,
Cytosol/Nucleus, Nucleus, Ribosome 60S, Ribosome 40S and ER), we want to
know how to optimally combine primary and auxiliary data. If we look at
figure @ref(fig:andypca), that illustrates the experimental separation
of the 10 spatial classes on a principal component plot, we see that
some organelles such as the mitochondrion or the cytosol and
cytosol/nucleus are well resolved, while others such as the Golgi or the
ER are less so. In this experiment, the former classes are not expected
to benefit from another data source, while the latter should benefit
from additional information.
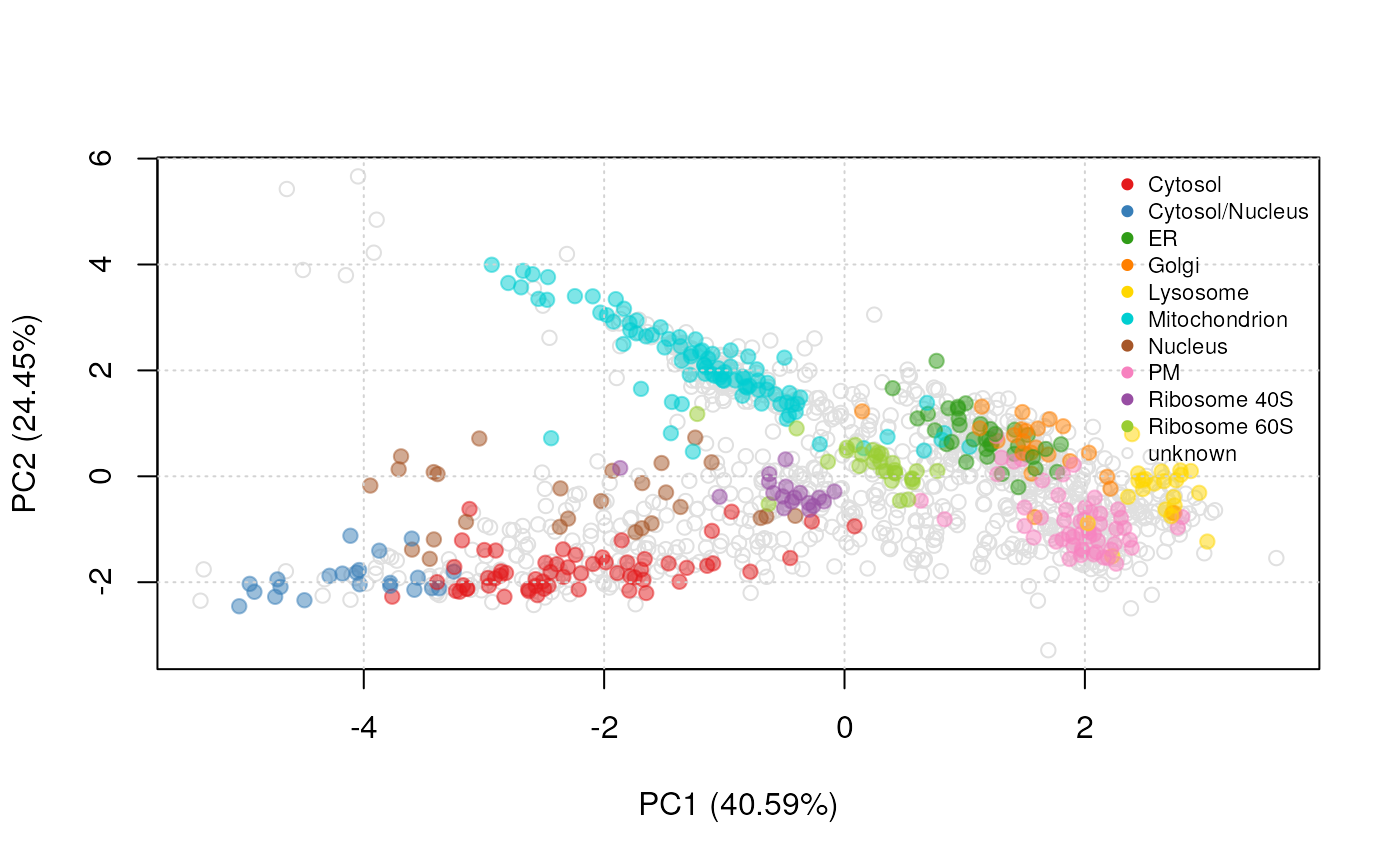
PCA plot of andy2011. The multivariate protein profiles are
summarised along the two first principal components. Proteins of unknown
localisation are represented by empty grey points. Protein markers,
which are well-known residents of specific sub-cellular niches are
colour-coded and form clusters on the figure.
Let’s define a set of three possible weights: 0, 0.5 and 1. A weight of 1 indicates that the final results rely exclusively on the experimental data and ignore completely the auxiliary data. A weight of 0 represents the opposite situation, where the primary data is ignored and only the auxiliary data is considered. A weight of 0.5 indicates that each data source will contribute equally to the final results. It is the algorithm’s optimisation step task to identify the optimal combination of class-specific weights for a given primary and auxiliary data pair. The optimisation process can be quite time consuming for many weights and many sub-cellular classes, as all combinations (there are possibilities; see below). One would generally defined more weights (for example 0, 0.25, 0.5, 0.75, 1 or 0, 0.33, 0.67, 1) to explore more fine-grained integration opportunities. The possible weight combinations can be calculated with the thetas function:
- 3 classes, 3 weights
## Weigths:
## (0, 0.5, 1)## [,1] [,2] [,3]
## [1,] 0 0.0 0.0
## [2,] 0 0.0 0.5
## [3,] 0 0.0 1.0
## [4,] 0 0.5 0.0
## [5,] 0 0.5 0.5
## [6,] 0 0.5 1.0## Weigths:
## (0, 0.5, 1)## [1] 27 3- 5 classes, 4 weights
## Weigths:
## (0, 0.333333333333333, 0.666666666666667, 1)## [1] 1024 5- for the human
andy2011data, considering 4 weights, there are very many combinations:
## marker classes for andy2011
m <- unique(fData(andy2011)$markers.tl)
m <- m[m != "unknown"]
th <- thetas(length(m), length.out=4)## Weigths:
## (0, 0.333333333333333, 0.666666666666667, 1)
dim(th)## [1] 1048576 10The actual combination of weights to be tested can be defined in
multiple ways: by passing a weights matrix explicitly (as those
generated with thetas above) through the th
argument; or by defining the increment between weights using
by; or by specifying the number of weights to be used
through the length.out argument.
Considering the sub-cellular resolution for this experiment, we would anticipate that the mitochondrion, the cytosol and the cytosol/nucleus classes would get high weights, while the ER and Golgi would be assigned lower weights.
As we use a nearest neighbour classifier, we also need to know how many neighbours to consider when classifying a protein of unknown localisation. The knnOptimisation function (see the pRoloc-tutorial vignette and the functions manual page) can be run on the primary and auxiliary data sources independently to estimate the best and values. Here, based on knnOptimisation, we use 3 and 3, for and respectively.
Finally, to assess the validity of the weight selection, it should be repeated a certain number of times (default value is 50). As the weight optimisation can become very time consuming for a wide range of weights and many target classes, we would recommend to start with a lower number of iterations, pre-analyse the results, proceed with further iterations and eventually combine the optimisation results data with the combineThetaRegRes function before proceeding with the selection of best weights.
topt <- knntlOptimisation(andy2011, andygoset,
th = th,
k = c(3, 3),
fcol = "markers.tl",
times = 50)The above code chunk would take too much time to be executed in the
frame of this vignette. Below, we pass a very small subset of theta
matrix to minimise the computation time. The knntlOptimisation
function supports parallelised execution using various backends thanks
to the BiocParallel
package; an appropriate backend will be defined automatically according
to the underlying architecture and user-defined backends can be defined
through the BPPARAM argument2. Also, in the interest
of time, the weights optimisation is repeated only 5 times below.
set.seed(1)
i <- sample(nrow(th), 12)
topt <- knntlOptimisation(andy2011, andygoset,
th = th[i, ],
k = c(3, 3),
fcol = "markers.tl",
times = 5)## Removing 501 columns with only 0s.## Note: vector will be ordered according to classes: Cytosol Cytosol/Nucleus ER Golgi Lysosome Mitochondrion Nucleus PM Ribosome 40S Ribosome 60S (as names are not explicitly defined)
topt## Object of class "ThetaRegRes"
## Algorithm: theta
## Theta hyper-parameters:
## weights: 0 0.3333333 0.6666667 1
## k: 3 3
## nrow: 12
## Design:
## Replication: 5 x 5-fold X-validation
## Partitioning: 0.2/0.8 (test/train)
## Results
## macro F1:
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.8096 0.8401 0.8812 0.8661 0.8869 0.9127
## best theta:
## Cytosol Cytosol.Nucleus ER Golgi Lysosome Mitochondrion Nucleus PM
## weight:0 0 0 5 4 0 0 1 0
## weight:0.33 0 4 0 0 0 1 0 4
## weight:0.67 1 1 0 1 0 0 0 0
## weight:1 4 0 0 0 5 4 4 1
## Ribosome.40S Ribosome.60S
## weight:0 5 0
## weight:0.33 0 4
## weight:0.67 0 1
## weight:1 0 0The optimisation is performed on the labelled marker examples only.
When removing unlabelled non-marker proteins (the
unknowns), some auxiliary GO columns end up containing only
0 (the GO-protein association was only observed in non-marker proteins),
which are temporarily removed.
The topt result stores all the result from the
optimisation step, and in particular the observed theta weights, which
can be directly plotted as shown on the bubble
plot below. These bubble plots give the proportion of best weights
for each marker class that was observed during the optimisation phase.
We see that the mitochondrion, the cytosol and cytosol/nucleus classes
predominantly are scored with height weights (2/3 and 1), consistent
with high reliability of the primary data. The Golgi and the ribosomal
clusters (and to a lesser extend the ER) favour smaller scores,
indicating a substantial benefit of the auxiliary data.
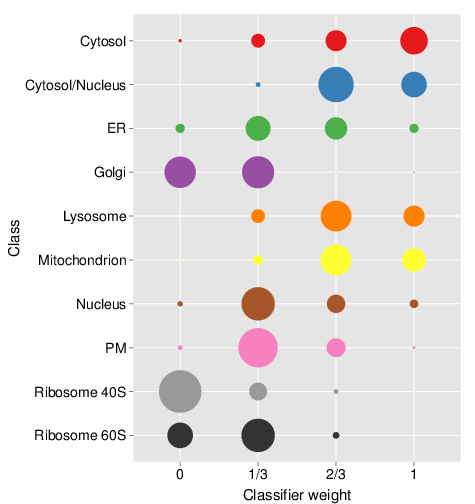
andy2011 and auxiliary
andygoset data sets, as produced by
plot(topt). This figure is not the result for the previous
code chunk, where only a random subset of 10 candidate weights have been
tested.Choosing weights
A set of best weights must be chosen and applied to the
classification of the unlabelled proteins (formally annotated as
unknown). These can be defined manually, based on the
pattern observed in the weights bubble plot,
or automatically extracted with the getParams method3. See
?getParams for details and the favourPrimary function,
if it is desirable to systematically favour the primary data (i.e. high
weights) when different weight combinations perform equally well.
getParams(topt)## Cytosol Cytosol/Nucleus ER Golgi Lysosome
## 1.0000000 0.3333333 0.0000000 0.0000000 1.0000000
## Mitochondrion Nucleus PM Ribosome 40S Ribosome 60S
## 1.0000000 1.0000000 0.3333333 0.0000000 0.3333333We provide the best parameters for the extensive parameter optimisation search, as provided by getParams:
(bw <- experimentData(andy2011)@other$knntl$thetas)## Cytosol Cytosol/Nucleus ER Golgi Lysosome
## 0.6666667 0.6666667 0.3333333 0.3333333 0.6666667
## Mitochondrion Nucleus PM Ribosome 40S Ribosome 60S
## 0.6666667 0.3333333 0.3333333 0.0000000 0.3333333Applying best theta weights
To apply our best weights and learn from the auxiliary data
accordingly when classifying the unlabelled proteins to one of the
sub-cellular niches considered in markers.tl (as displayed
on figure @ref(fig:andypca)), we pass the primary and auxiliary data
sets, best weights, best k’s (and, on our case the marker’s feature
variable we want to use, default would be markers) to the
knntlClassification function.
andy2011 <- knntlClassification(andy2011, andygoset,
bestTheta = bw,
k = c(3, 3),
fcol = "markers.tl")This will generate a new instance of class MSnSet, identical
to the primary data, including the classification results and
classifications scores of the transfer learning classification algorithm
(as knntl and knntl.scores feature variables
respectively). Below, we extract the former with the
getPrediction function and plot the results of the
classification.
andy2011 <- getPredictions(andy2011, fcol = "knntl")## ans
## Chromatin associated Cytosol Cytosol/Nucleus
## 11 293 43
## Endosome ER Golgi
## 12 194 76
## Lysosome Mitochondrion Nucleus
## 64 260 110
## PM Ribosome 40S Ribosome 60S
## 234 19 55
setStockcol(paste0(getStockcol(), "80"))
ptsze <- exp(fData(andy2011)$knntl.scores) - 1
plot2D(andy2011, fcol = "knntl", cex = ptsze)
setStockcol(NULL)
addLegend(andy2011, where = "topright",
fcol = "markers.tl",
bty = "n", cex = .7)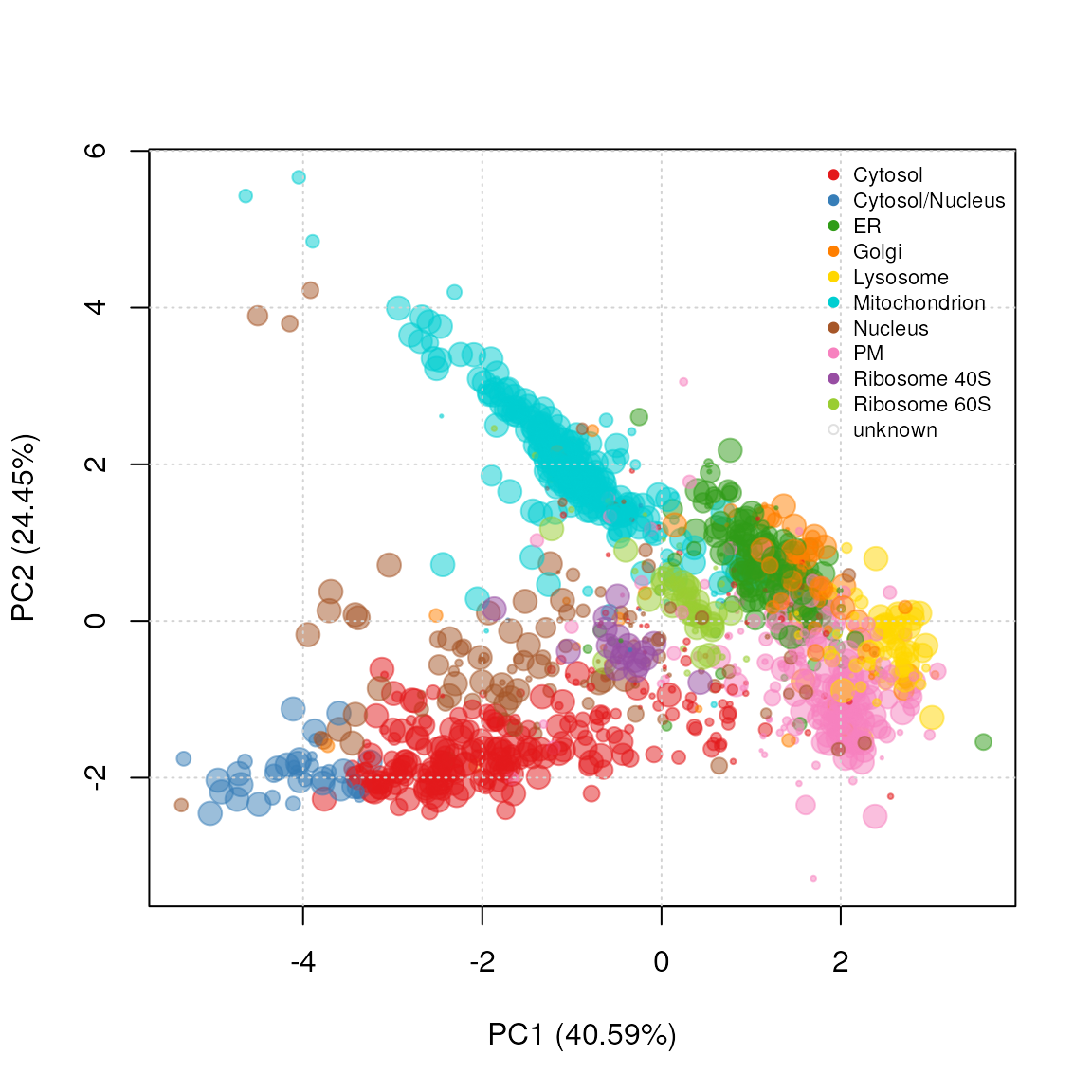
PCA plot of andy2011 after transfer learning
classification. The size of the points is proportional to the
classification scores.
Please read the pRoloc-tutorial vignette, and in particular the classification section, for more details on how to proceed with exploration the classification results and classification scores.
Conclusions
This vignette describes the application of a weighted -nearest neighbour transfer learning algorithm and its application to the sub-cellular localisation prediction of proteins using quantitative proteomics data as primary data and Gene Ontology-derived binary data as auxiliary data source. The algorithm can be used with various data sources (we show how to compile binary data from the Human Protein Atlas in section @ref(sec:hpaaux)) and have successfully applied the algorithm (L. M. Breckels et al. 2016) on third-party quantitative auxiliary data.
Session information
All software and respective versions used to produce this document are listed below.
## R Under development (unstable) (2024-11-20 r87352)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.1 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] hpar_1.49.0 biomaRt_2.63.0 class_7.3-22
## [4] pRolocdata_1.45.1 pRoloc_1.47.1 BiocParallel_1.41.0
## [7] MLInterfaces_1.87.0 cluster_2.1.6 annotate_1.85.0
## [10] XML_3.99-0.17 AnnotationDbi_1.69.0 IRanges_2.41.1
## [13] MSnbase_2.33.2 ProtGenerics_1.39.0 S4Vectors_0.45.2
## [16] mzR_2.41.1 Rcpp_1.0.13-1 Biobase_2.67.0
## [19] BiocGenerics_0.53.3 generics_0.1.3 knitr_1.49
## [22] BiocStyle_2.35.0
##
## loaded via a namespace (and not attached):
## [1] splines_4.5.0 filelock_1.0.3
## [3] tibble_3.2.1 hardhat_1.4.0
## [5] preprocessCore_1.69.0 pROC_1.18.5
## [7] rpart_4.1.23 lifecycle_1.0.4
## [9] httr2_1.0.6 doParallel_1.0.17
## [11] globals_0.16.3 lattice_0.22-6
## [13] MASS_7.3-61 MultiAssayExperiment_1.33.1
## [15] dendextend_1.19.0 magrittr_2.0.3
## [17] limma_3.63.2 plotly_4.10.4
## [19] sass_0.4.9 rmarkdown_2.29
## [21] jquerylib_0.1.4 yaml_2.3.10
## [23] MsCoreUtils_1.19.0 DBI_1.2.3
## [25] RColorBrewer_1.1-3 lubridate_1.9.3
## [27] abind_1.4-8 zlibbioc_1.53.0
## [29] GenomicRanges_1.59.1 purrr_1.0.2
## [31] mixtools_2.0.0 AnnotationFilter_1.31.0
## [33] nnet_7.3-19 rappdirs_0.3.3
## [35] ipred_0.9-15 lava_1.8.0
## [37] GenomeInfoDbData_1.2.13 listenv_0.9.1
## [39] parallelly_1.39.0 pkgdown_2.1.1.9000
## [41] ncdf4_1.23 codetools_0.2-20
## [43] DelayedArray_0.33.2 xml2_1.3.6
## [45] tidyselect_1.2.1 UCSC.utils_1.3.0
## [47] viridis_0.6.5 matrixStats_1.4.1
## [49] BiocFileCache_2.15.0 jsonlite_1.8.9
## [51] caret_6.0-94 e1071_1.7-16
## [53] survival_3.7-0 iterators_1.0.14
## [55] systemfonts_1.1.0 foreach_1.5.2
## [57] segmented_2.1-3 tools_4.5.0
## [59] progress_1.2.3 ragg_1.3.3
## [61] glue_1.8.0 prodlim_2024.06.25
## [63] gridExtra_2.3 SparseArray_1.7.2
## [65] xfun_0.49 MatrixGenerics_1.19.0
## [67] GenomeInfoDb_1.43.1 dplyr_1.1.4
## [69] withr_3.0.2 BiocManager_1.30.25
## [71] fastmap_1.2.0 fansi_1.0.6
## [73] digest_0.6.37 mime_0.12
## [75] timechange_0.3.0 R6_2.5.1
## [77] textshaping_0.4.0 colorspace_2.1-1
## [79] gtools_3.9.5 lpSolve_5.6.22
## [81] RSQLite_2.3.8 utf8_1.2.4
## [83] tidyr_1.3.1 hexbin_1.28.5
## [85] data.table_1.16.2 recipes_1.1.0
## [87] FNN_1.1.4.1 prettyunits_1.2.0
## [89] PSMatch_1.11.0 httr_1.4.7
## [91] htmlwidgets_1.6.4 S4Arrays_1.7.1
## [93] ModelMetrics_1.2.2.2 pkgconfig_2.0.3
## [95] gtable_0.3.6 timeDate_4041.110
## [97] blob_1.2.4 impute_1.81.0
## [99] XVector_0.47.0 htmltools_0.5.8.1
## [101] bookdown_0.41 MALDIquant_1.22.3
## [103] clue_0.3-66 scales_1.3.0
## [105] png_0.1-8 gower_1.0.1
## [107] reshape2_1.4.4 coda_0.19-4.1
## [109] nlme_3.1-166 curl_6.0.1
## [111] proxy_0.4-27 cachem_1.1.0
## [113] stringr_1.5.1 BiocVersion_3.21.1
## [115] parallel_4.5.0 mzID_1.45.0
## [117] vsn_3.75.0 desc_1.4.3
## [119] pillar_1.9.0 grid_4.5.0
## [121] vctrs_0.6.5 pcaMethods_1.99.0
## [123] randomForest_4.7-1.2 dbplyr_2.5.0
## [125] xtable_1.8-4 evaluate_1.0.1
## [127] mvtnorm_1.3-2 cli_3.6.3
## [129] compiler_4.5.0 rlang_1.1.4
## [131] crayon_1.5.3 future.apply_1.11.3
## [133] LaplacesDemon_16.1.6 mclust_6.1.1
## [135] QFeatures_1.17.0 affy_1.85.0
## [137] plyr_1.8.9 fs_1.6.5
## [139] stringi_1.8.4 viridisLite_0.4.2
## [141] munsell_0.5.1 Biostrings_2.75.1
## [143] lazyeval_0.2.2 Matrix_1.7-1
## [145] ExperimentHub_2.15.0 hms_1.1.3
## [147] bit64_4.5.2 future_1.34.0
## [149] ggplot2_3.5.1 KEGGREST_1.47.0
## [151] statmod_1.5.0 AnnotationHub_3.15.0
## [153] SummarizedExperiment_1.37.0 kernlab_0.9-33
## [155] igraph_2.1.1 memoise_2.0.1
## [157] affyio_1.77.0 bslib_0.8.0
## [159] sampling_2.10 bit_4.5.0